Basic Usage, small protein
Gromacs_py basic example
Here is an example of a short simulation of the SH3 domain of phospholipase C$:nbsphinx-math:`gamma`$1. Five successive steps are used:
Topologie creation using
GmxSys.prepare_top().Minimisation of the structure using
GmxSys.em_2_steps().Solvation of the system using
GmxSys.solvate_add_ions().Equilibration of the system using
GmxSys.em_equi_three_step_iter_error().Production run using
GmxSys.production().
Import
[1]:
import sys
import os
import urllib.request
import pandas as pd
import matplotlib
import matplotlib.pyplot as plt
import numpy as np
import seaborn as sns
To use
gromacs_pyin a project:
[2]:
from gromacs_py import gmx
Simulation setup
Define a few variables for you simulation, like:
simulation output folders
ionic concentration
number of minimisation steps
equilibration and production time
[3]:
DATA_OUT = 'data_sim'
PDB_ID = '1Y0M'
# System Setup
vsite='none'
ion_C = 0.15
sys_top_folder = os.path.join(DATA_OUT, 'sys_top')
# Energy Minimisation
em_folder = os.path.join(DATA_OUT, 'em')
em_sys_folder = os.path.join(DATA_OUT, 'sys_em')
em_step_number = 5000
# Equillibration
equi_folder = os.path.join(DATA_OUT, 'sys_equi')
HA_time = 0.5
CA_time = 1.0
CA_LOW_time = 2.0
dt_HA = 0.001
dt = 0.002
HA_step = 1000 * HA_time / dt_HA
CA_step = 1000 * CA_time / dt
CA_LOW_step = 1000 * CA_LOW_time / dt
# Production
prod_folder = os.path.join(DATA_OUT, 'sys_prod')
prod_time = 10.0
prod_step = 1000 * prod_time / dt
Get PDB file from the rcsb.org website
[4]:
os.makedirs(DATA_OUT, exist_ok = True)
raw_pdb = urllib.request.urlretrieve('http://files.rcsb.org/download/{}.pdb'.format(PDB_ID),
'{}/{}.pdb'.format(DATA_OUT, PDB_ID))
Create the GmxSys object
[5]:
md_sys = gmx.GmxSys(name=PDB_ID, coor_file=raw_pdb[0])
md_sys.display()
name : 1Y0M
coor_file : data_sim/1Y0M.pdb
nt : 0
ntmpi : 0
sys_history : 0
Create topology:
Topologie creation involves: - protonation calculation using pdb2pqr and propka - topologie creation using pdb2gmx
[6]:
md_sys.prepare_top(out_folder=os.path.join(DATA_OUT, 'prot_top'), vsite=vsite, ff='amber99sb-ildn')
md_sys.create_box(dist=1.0, box_type="dodecahedron", check_file_out=True)
3D coordinates vizualisation using nglview
Use the view_coor() function of the GmxSys object to vizualise the protein coordinates.
Note that nglview library need to be installed :
conda install nglview
You’ll probably need to restart you notebook after installation to enable nglview widget appearance.
[7]:
view = md_sys.view_coor()
view.add_representation(repr_type='licorice', selection='protein')
view
[9]:
# Unecessary, only need to nglview online:
IFrame(src='../_static/1Y0M.html', width=800, height=300)
[9]:
Energy minimisation
[12]:
md_sys.em_2_steps(out_folder=em_folder,
no_constr_nsteps=em_step_number,
constr_nsteps=em_step_number,
posres="",
create_box_flag=False,
emtol=0.1, nstxout=100)
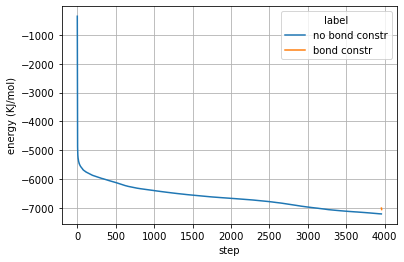
Plot energy:
[30]:
ener_pd_1 = md_sys.sys_history[-1].get_ener(selection_list=['Potential'])
ener_pd_2 = md_sys.get_ener(selection_list=['Potential'])
ener_pd_1['label'] = 'no bond constr'
ener_pd_2['label'] = 'bond constr'
ener_pd = pd.concat([ener_pd_1, ener_pd_2])
ener_pd['Time (ps)'] = np.arange(len(ener_pd))
gmx energy -f data_sim/em/Init_em_1Y0M.edr -o tmp_edr.xvg
gmx energy -f data_sim/em/1Y0M.edr -o tmp_edr.xvg
[31]:
ax = sns.lineplot(x="Time (ps)", y="Potential",
hue="label",
data=ener_pd)
ax.set_xlabel('step')
ax.set_ylabel('energy (KJ/mol)')
plt.grid()
3D vizualisation using nglview
Not much append in the second minimisation, we can have a look at the first one using md_sys.sys_history[-1], which is considered as a GmxSys object.
Use the coor_traj atribute of the GmxSys object to vizualise the trajectory. Note that that the simpletraj library is a dependenie. To install simpletraj use:
pip install simpletraj
first you should make molecule whole using
convert_trj()function.
[14]:
md_sys.sys_history[-1].convert_trj()
[75]:
view = md_sys.sys_history[-1].view_traj()
view.add_representation(repr_type='licorice', selection='protein')
view.center()
view
[77]:
# Unecessary, only need to nglview online:
IFrame(src='../_static/1Y0M_em_traj.html', width=800, height=300)
[77]:
Solvation (water and \(Na^{+} Cl^{-}\))
[18]:
md_sys.solvate_add_ions(out_folder=sys_top_folder,
ion_C=ion_C)
md_sys.display()
name : 1Y0M
sim_name : 1Y0M
coor_file : data_sim/sys_top/1Y0M_water_ion.gro
top_file : data_sim/sys_top/1Y0M_water_ion.top
tpr : data_sim/em/1Y0M.tpr
mdp : data_sim/em/1Y0M.mdp
xtc : data_sim/em/1Y0M.trr
edr : data_sim/em/1Y0M.edr
log : data_sim/em/1Y0M.log
nt : 0
ntmpi : 0
sys_history : 4
System minimisation and equilibration
[19]:
md_sys.em_equi_three_step_iter_error(out_folder=equi_folder,
no_constr_nsteps=em_step_number,
constr_nsteps=em_step_number,
nsteps_HA=HA_step,
nsteps_CA=CA_step,
nsteps_CA_LOW=CA_LOW_step,
dt=dt, dt_HA=dt_HA,
vsite=vsite, maxwarn=1)
gmx mdrun -s equi_HA_1Y0M.tpr -deffnm equi_HA_1Y0M -nt 0 -ntmpi 0 -nsteps -2 -nocopyright -append -cpi equi_HA_1Y0M.cpt
gmx grompp -f equi_CA_1Y0M.mdp -c ../00_equi_HA/equi_HA_1Y0M.gro -r ../../sys_em/1Y0M_compact.pdb -p ../../../sys_top/1Y0M_water_ion.top -po out_equi_CA_1Y0M.mdp -o equi_CA_1Y0M.tpr -maxwarn 1
gmx mdrun -s equi_CA_1Y0M.tpr -deffnm equi_CA_1Y0M -nt 0 -ntmpi 0 -nsteps -2 -nocopyright
gmx grompp -f equi_CA_LOW_1Y0M.mdp -c ../01_equi_CA/equi_CA_1Y0M.gro -r ../../sys_em/1Y0M_compact.pdb -p ../../../sys_top/1Y0M_water_ion.top -po out_equi_CA_LOW_1Y0M.mdp -o equi_CA_LOW_1Y0M.tpr -maxwarn 1
gmx mdrun -s equi_CA_LOW_1Y0M.tpr -deffnm equi_CA_LOW_1Y0M -nt 0 -ntmpi 0 -nsteps -2 -nocopyright
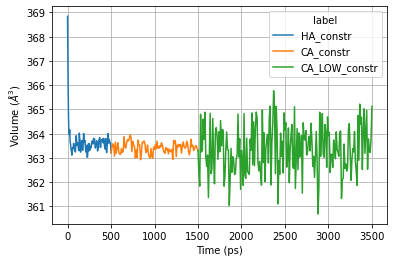
Plot temperature
[35]:
ener_pd_1 = md_sys.sys_history[-2].get_ener(selection_list=['Volume'])
ener_pd_2 = md_sys.sys_history[-1].get_ener(selection_list=['Volume'])
ener_pd_3 = md_sys.get_ener(selection_list=['Volume'])
ener_pd_1['label'] = 'HA_constr'
ener_pd_2['label'] = 'CA_constr'
ener_pd_2['Time (ps)'] = ener_pd_2['Time (ps)'] + ener_pd_1['Time (ps)'].max()
ener_pd_3['label'] = 'CA_LOW_constr'
ener_pd_3['Time (ps)'] = ener_pd_3['Time (ps)'] + ener_pd_2['Time (ps)'].max()
ener_pd = pd.concat([ener_pd_1, ener_pd_2, ener_pd_3])
gmx energy -f data_sim/sys_equi/sys_equi/00_equi_HA/equi_HA_1Y0M.edr -o tmp_edr.xvg
gmx energy -f data_sim/sys_equi/sys_equi/01_equi_CA/equi_CA_1Y0M.edr -o tmp_edr.xvg
gmx energy -f data_sim/sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.edr -o tmp_edr.xvg
[36]:
ax = sns.lineplot(x="Time (ps)", y="Volume",
hue="label",
data=ener_pd)
ax.set_ylabel('Volume ($Å^3$)')
plt.grid()
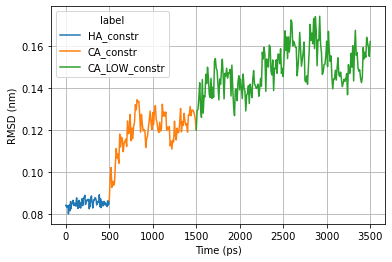
Plot RMSD
[42]:
# Define reference structure for RMSD calculation
ref_sys = md_sys.sys_history[1]
rmsd_pd_1 = md_sys.sys_history[-2].get_rmsd(['C-alpha', 'Protein'], ref_sys=ref_sys)
rmsd_pd_2 = md_sys.sys_history[-1].get_rmsd(['C-alpha', 'Protein'], ref_sys=ref_sys)
rmsd_pd_3 = md_sys.get_rmsd(['C-alpha', 'Protein'], ref_sys=ref_sys)
rmsd_pd_1['label'] = 'HA_constr'
rmsd_pd_2['label'] = 'CA_constr'
rmsd_pd_2['time'] = rmsd_pd_2['time'] + rmsd_pd_1['time'].max()
rmsd_pd_3['label'] = 'CA_LOW_constr'
rmsd_pd_3['time'] = rmsd_pd_3['time'] + rmsd_pd_2['time'].max()
rmsd_pd = pd.concat([rmsd_pd_1, rmsd_pd_2, rmsd_pd_3])
gmx rms -s data_sim/em/Init_em_1Y0M.tpr -f data_sim/sys_equi/sys_equi/00_equi_HA/equi_HA_1Y0M.xtc -n data_sim/sys_equi/sys_equi/00_equi_HA/equi_HA_1Y0M.ndx -o tmp_rmsd.xvg -fit rot+trans -ng 1 -pbc no
gmx rms -s data_sim/em/Init_em_1Y0M.tpr -f data_sim/sys_equi/sys_equi/01_equi_CA/equi_CA_1Y0M.xtc -n data_sim/sys_equi/sys_equi/01_equi_CA/equi_CA_1Y0M.ndx -o tmp_rmsd.xvg -fit rot+trans -ng 1 -pbc no
gmx rms -s data_sim/em/Init_em_1Y0M.tpr -f data_sim/sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.xtc -n data_sim/sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.ndx -o tmp_rmsd.xvg -fit rot+trans -ng 1 -pbc no
[43]:
ax = sns.lineplot(x="time", y="Protein",
hue="label",
data=rmsd_pd)
ax.set_ylabel('RMSD (nm)')
ax.set_xlabel('Time (ps)')
plt.grid()
Production
[27]:
md_sys.production(out_folder=prod_folder,
nsteps=prod_step,
dt=dt, vsite=vsite, maxwarn=1)
gmx grompp -f prod_1Y0M.mdp -c ../sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.gro -r ../sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.gro -p ../sys_top/1Y0M_water_ion.top -po out_prod_1Y0M.mdp -o prod_1Y0M.tpr -maxwarn 1 -n ../sys_equi/sys_equi/02_equi_CA_LOW/equi_CA_LOW_1Y0M.ndx
gmx mdrun -s prod_1Y0M.tpr -deffnm prod_1Y0M -nt 0 -ntmpi 0 -nsteps -2 -nocopyright
Prepare trajectory
[40]:
# Center trajectory
md_sys.center_mol_box(traj=True)
gmx make_ndx -f data_sim/sys_prod/prod_1Y0M.gro -o data_sim/sys_prod/prod_1Y0M.ndx
gmx trjconv -f data_sim/sys_prod/prod_1Y0M.xtc -o data_sim/sys_prod/prod_1Y0M_compact.xtc -s data_sim/sys_prod/prod_1Y0M.tpr -ur tric -pbc mol -center yes -n data_sim/sys_prod/prod_1Y0M.ndx
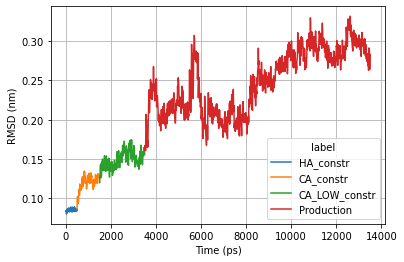
Basic Analysis
[44]:
rmsd_prod_pd = md_sys.get_rmsd(['C-alpha', 'Protein'], ref_sys=ref_sys)
rmsd_prod_pd['label'] = 'Production'
rmsd_prod_pd['time'] = rmsd_prod_pd['time'] + rmsd_pd['time'].max()
rmsd_all_pd = pd.concat([rmsd_pd, rmsd_prod_pd])
gmx rms -s data_sim/em/Init_em_1Y0M.tpr -f data_sim/sys_prod/prod_1Y0M_compact.xtc -n data_sim/sys_prod/prod_1Y0M.ndx -o tmp_rmsd.xvg -fit rot+trans -ng 1 -pbc no
[48]:
ax = sns.lineplot(x="time", y="Protein",
hue="label",
data=rmsd_all_pd)
ax.set_ylabel('RMSD (nm)')
ax.set_xlabel('Time (ps)')
plt.grid()
Trajectory vizualisation
[58]:
# Align the protein coordinates
md_sys.convert_trj(select='Protein\nSystem\n', fit='rot+trans', pbc='none', skip='10')
gmx trjconv -f data_sim/sys_prod/prod_1Y0M_compact_compact.xtc -o data_sim/sys_prod/prod_1Y0M_compact_compact_compact.xtc -s data_sim/sys_prod/prod_1Y0M.tpr -ur compact -pbc none -fit rot+trans -n data_sim/sys_prod/prod_1Y0M.ndx -skip 10
[70]:
view = md_sys.view_traj()
view.add_representation(repr_type='licorice', selection='protein')
view.center(selection='CA')
view
[4]:
# Unecessary, only need to nglview online:
IFrame(src='../_static/1Y0M_prod_traj.html', width=800, height=300)
[4]:
[ ]: